Introduction
|
Alexander disease is a rare, fatal neuro-degenerative disorder characterized by the presence of Rosenthal fibers in specialized brain cells called astrocytes [2,6]. This disease is caused by gain of function mutations in the gene GFAP, an intermediate filament protein, which is primarily expressed in astrocytes. These mutations to GFAP result in the over-expression and accumulation of mutated GFAP aggregates, which makeup Rosenthal fibers [2]. There are currently four types of Alexander Disease which are recognized based on age of onset, including: neonatal, infantile, juvenile, and adult forms. Symptoms will vary, but can include neuronal dysfunction, seizures, and developmental defects [2,5,6]. Severity decreases with increased age of onset, as neonatal and infantile cases are usually fatal within 2 years, whereas juvenile and adult cases are usually fatal within 20 years [2,5].
In the past, research has shown that astrocytes are important regulators of extracellular neurotransmitter levels within the brain [3]. In Alexander disease, GFAP mutant astrocytes are no longer able to efficiently uptake extracellular glutamate, resulting in toxic glutamate levels, the destruction of neurons, and neuronal dysfunction [1,3]. Although these studies implicate GFAP in glutamate regulation and transport in astrocytes, the cellular mechanisms of how GFAP performs these functions is unclear. |

Figure 1: Effect of GFAP mutations on glutamate uptake.
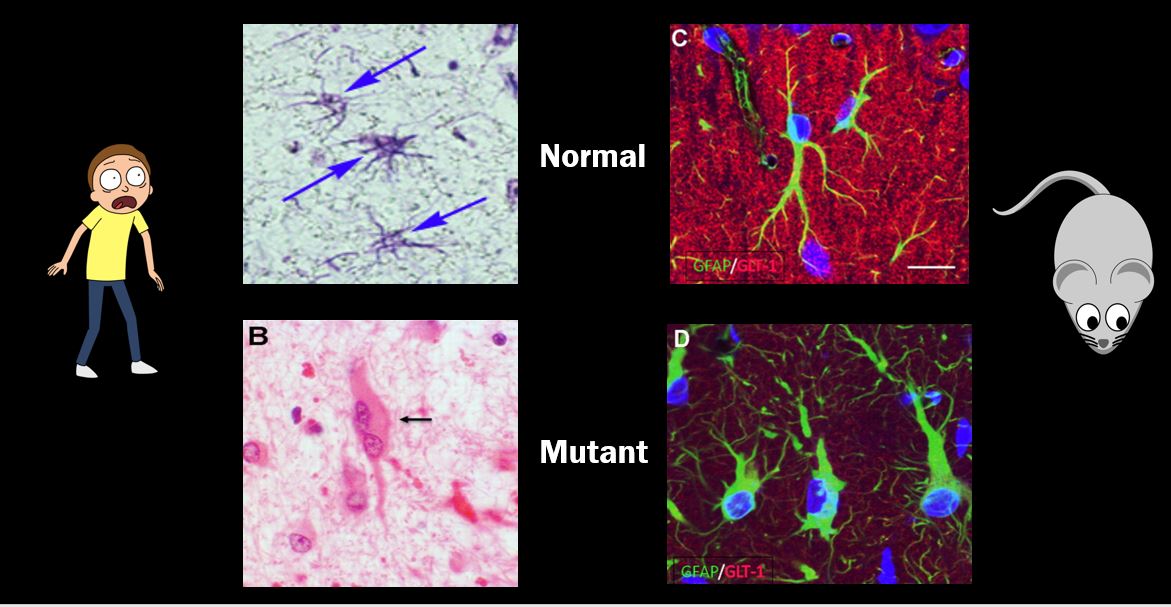
Figure 2: Human and Mouse normal and mutant astrocytes.
|
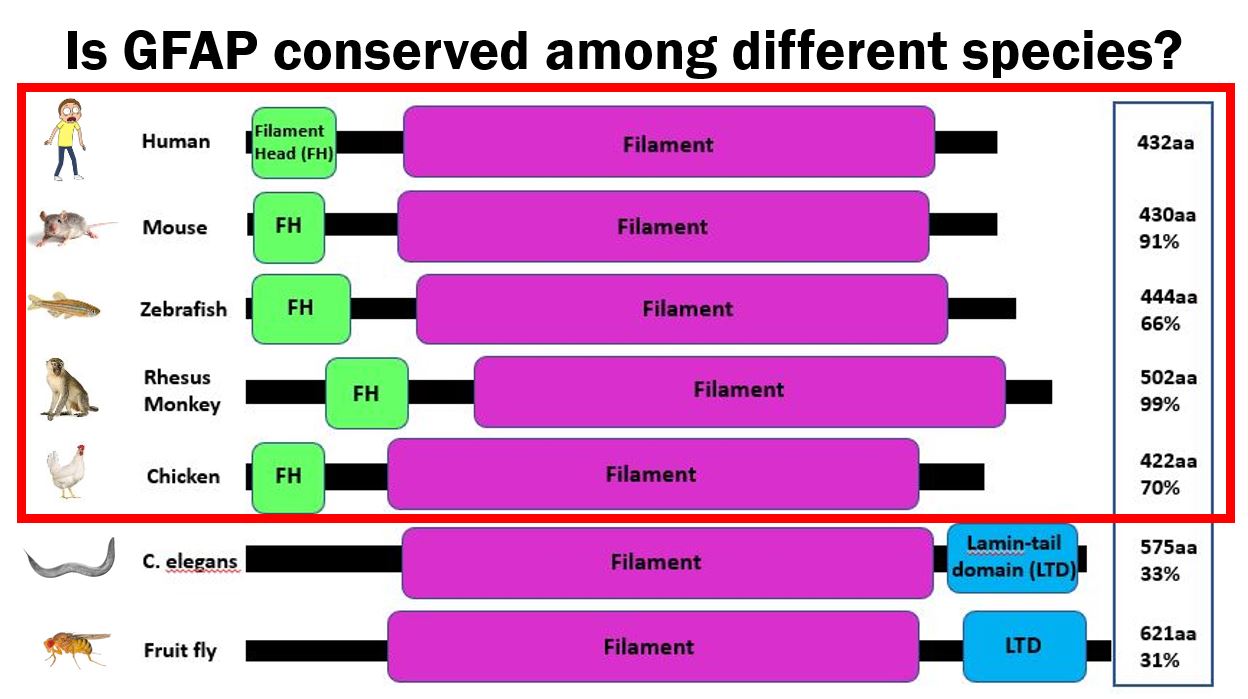
Given GFAP's implicated role in glutamate uptake and neuronal regulation in human astrocytes, I expect GFAP to play a similar role in organisms who share GFAP homology and have similar astrocyte biology. Looking at Figures 3 and 4, human GFAP and its two protein domains are closely conserved among vertebrates, with an especially high similarity to mice (Mus musculus). Due to the similarity between mice and human GFAP homologs, as well as the similar Alexander disease phenotype seen in Figure 2, I chose the mouse as my model organism. My primary goal is to determine the role of GFAP in glutamate uptake and neuronal regulation in astrocytes, and I hypothesize that GFAP plays a role in mediating these twp functions.
|

|
|
Figure 3: Conserved domains of GFAP.
|
Figure 4: Maximum likelihood phylogeny of GFAP homologs.
|
Specific Aim 1
In my first aim, I intend to identify conserved amino acids in GFAP, which are important for glutamate uptake and neuronal regulation. To do this, I will perform a multiple sequence alignment in MEGA using all GFAP homologs which have been identified by BLAST. I will then look for amino acids which are conserved (Figure 5), and use CRISPR/Cas9 to knockout the conserved sites in mouse embryos to create mutants. Following this, I will sacrifice the wild type and mutant mice, and sample their white matter, as this area is dense with astrocytes. With the samples, I will look for classic signs of Alexander disease, such as Rosenthal fibers, and I will culture the astrocytes to perform a glutamate uptake assay to determine whether or not glutamate uptake and neuronal regulation are inhibited (Figure 6) [4]. I expect to see reduced glutamate uptake in the mutants compared to the wild type mice.

Figure 5: Multiple sequence alignment to identify conserved sites among all GFAP homologs.
|
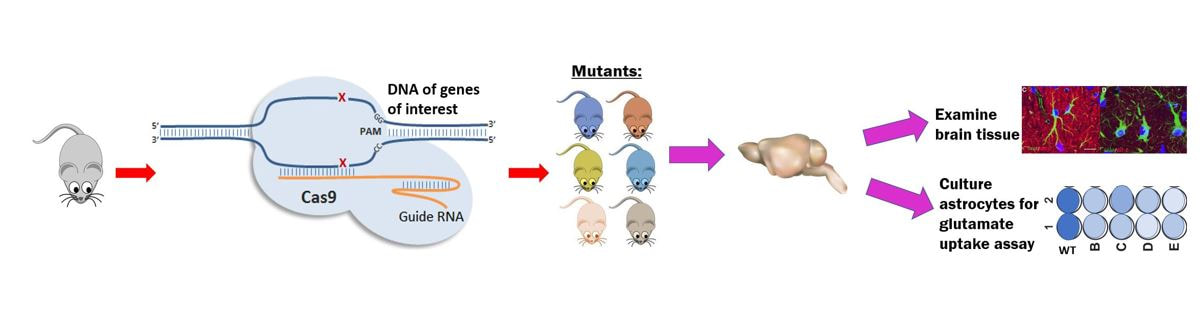
Figure 6: General walk through of CRISPR knockouts to glutamate assay which is utilized in all aims.
|
Specific Aim 2
For my second aim, I will identify differentially expressed genes in the mutant mice from Aim 1. To do this, I will perform a RNA-seq analysis on the brain tissue of wild type and mutant mice. I will then use Gene Ontology to sort through the differentially expressed genes in order to find which ones are implicated to be involved in glutamate uptake and neuronal regulation (Figure 7). I will then repeat the general work flow outlined in Figure 6, where I will knock out the differentially expressed genes involved in glutamate uptake and neuronal regulation, and observe whether or not these mutations actually result in these functions and have a similar astrocyte biology as seen in Alexander disease. I expect to find new differentially expressed genes which are involved in glutamate uptake and neuronal regulation in astrocytes.

|

|
Figure 7: Hypothetical walk through of identifying deferentially expressed genes in GFAP mutant mice and determining their function in Gene Ontology.
Specific Aim 3
For my final aim, I will identify new protein interactors of GFAP. To do this I will use Tap-MS screening in wild type mice, and tag the N-terminus of GFAP to maintain the proteins' natural function. The interacting proteins will be obtained by affinity purification and mass spectrometry, as seen in Figure 8. I will then use Gene Ontology to sort through the genes of my interacting proteins in order to find which genes are implicated to be involved in glutamate uptake and neuronal regulation. The genes of the interacting proteins will be knocked out following Figure 6, and I will observe whether or not these mutations result in a similar astrocyte biology as seen in Alexander disease. I will also confirm whether or not these genes play a role in glutamate uptake and neuronal regulation through the glutamate assay. I expect to find new protein interactors which are involved these functions.

|

|
Figure 8: Hypothetical walk through identifying new protein interactions through Tap-MS and determining their role through Gene Ontology.

|
Conclusions and Future DirectionsBy elucidating the cellular mechanisms behind how GFAP functions in glutamate uptake and neuronal regulation in astrocytes, it may be possible to understand how GFAP functions in other cellular processes within the brain and the uptake of other neurotransmitters. Furthermore, it may be possible to find future treatments which can help restore functional neurotransmitter uptake. For future directions, I would like to do a chemical screening on cultured mutant astrocytes to see if I could restore functional glutamate uptake in hopes for the development of a therapeutic treatment for juvenile and adult patients. Another direction I would possibly pursue, would be to do a chemical screening to find drugs which result in lower accumulations of Rosenthal fibers, as it is possible the burden of these aggregates may play a role in the astrocytes' diseased functions.
|
Draft and Final Presentations
|
| ||||
References
[1] Liu, Y.P., Yang, C. S., & Tzeng, S. F. (2007). Inhibitory regulation of glutamate aspartate transporter (GLAST) expression in astrocytes by cadmium‐induced calcium influx. Journal of Neurochemistry. Wiley Online Library. Retrieved May 13, 2018, from https://onlinelibrary.wiley.com/doi/abs/10.1111/j.1471-4159.2007.05118.x
[2] Johnson, A. B., & Brenner, M. (2003). Alexander’s Disease: Clinical, Pathologic, and Genetic Features. Journal of Child Neurology, 18(9), 625–632.
[3] Olabarria, M., & Goldman, J. E. (2017). Disorders of Astrocytes: Alexander Disease as a Model. Annual Review of Pathology: Mechanisms of Disease, 12(1), 131–152. https://doi.org/10.1146/annurev-pathol-052016-100218
[4] Spanagel, R., Pendyala, G., Abarca, C., Zghoul, T., Sanchis-Segura, C., Magnone, M. C., … Albrecht, U. (2005). ERRATUM: The clock gene Per2 influences the glutamatergic system and modulates alcohol consumption. Nature Medicine, 11(2), 233. https://doi.org/10.1038/nm0205-233c
[5] Springer, S., Erlewein, R., Naegele, T., Becker, I., Auer, D., Grodd, W., & Krägeloh-Mann, I. (2000). Alexander disease--classification revisited and isolation of a neonatal form. Neuropediatrics, 31(2), 86–92. https://doi.org/10.1055/s-2000-7479
[6] Srivastava, S., & Naidu, S. (1993). Alexander Disease. In M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. Bean, K. Stephens, & A. Amemiya (Eds.), GeneReviews®. Seattle (WA): University of Washington, Seattle. Retrieved from http://www.ncbi.nlm.nih.gov/books/NBK1172/
[2] Johnson, A. B., & Brenner, M. (2003). Alexander’s Disease: Clinical, Pathologic, and Genetic Features. Journal of Child Neurology, 18(9), 625–632.
[3] Olabarria, M., & Goldman, J. E. (2017). Disorders of Astrocytes: Alexander Disease as a Model. Annual Review of Pathology: Mechanisms of Disease, 12(1), 131–152. https://doi.org/10.1146/annurev-pathol-052016-100218
[4] Spanagel, R., Pendyala, G., Abarca, C., Zghoul, T., Sanchis-Segura, C., Magnone, M. C., … Albrecht, U. (2005). ERRATUM: The clock gene Per2 influences the glutamatergic system and modulates alcohol consumption. Nature Medicine, 11(2), 233. https://doi.org/10.1038/nm0205-233c
[5] Springer, S., Erlewein, R., Naegele, T., Becker, I., Auer, D., Grodd, W., & Krägeloh-Mann, I. (2000). Alexander disease--classification revisited and isolation of a neonatal form. Neuropediatrics, 31(2), 86–92. https://doi.org/10.1055/s-2000-7479
[6] Srivastava, S., & Naidu, S. (1993). Alexander Disease. In M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. Bean, K. Stephens, & A. Amemiya (Eds.), GeneReviews®. Seattle (WA): University of Washington, Seattle. Retrieved from http://www.ncbi.nlm.nih.gov/books/NBK1172/
