What is a Phylogeny?
|
Phylogenetics is the study of the evolutionary relationships between different species over time. Phylogenetics stems from the theory that all organisms share a single common ancestor, so therefore all genes and their corresponding proteins that are present now have been passed on from a most recent common ancestor [2]! A Phylogeny is a visual explanation of the evolutionary relationships between different genes and proteins of different species [2]. Phylogenies are typically found in the form of a tree, called a phylogenetic tree. The Root represents the most recent common ancestor of all listed organisms, while a node represents the most recent common ancestor of two or more species. Each node represents a new speciation event, and clades represent groups of closely related organisms . Branch lengths depict evolutionary time and accumulations of new mutations (Figure 1).
|
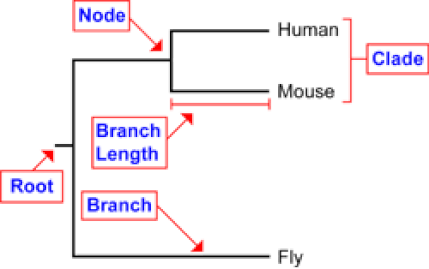
Figure 1: Diagram which illustrates the important parts of phylogenetic trees.
|
What is Clustal Omega?
Clustal Omega is a bioinformatic program which allows researchers to align many selected sequences in order to determine the evolutionary relationship between protein homologs. These alignments serve useful in discovering conserved domains, and understanding how different speciation events affected the protein structure over time [3,4]. For the trees below, Clustal Omega was used to align 17 sequences and aligned sequences were then processed into trees using MEGA.
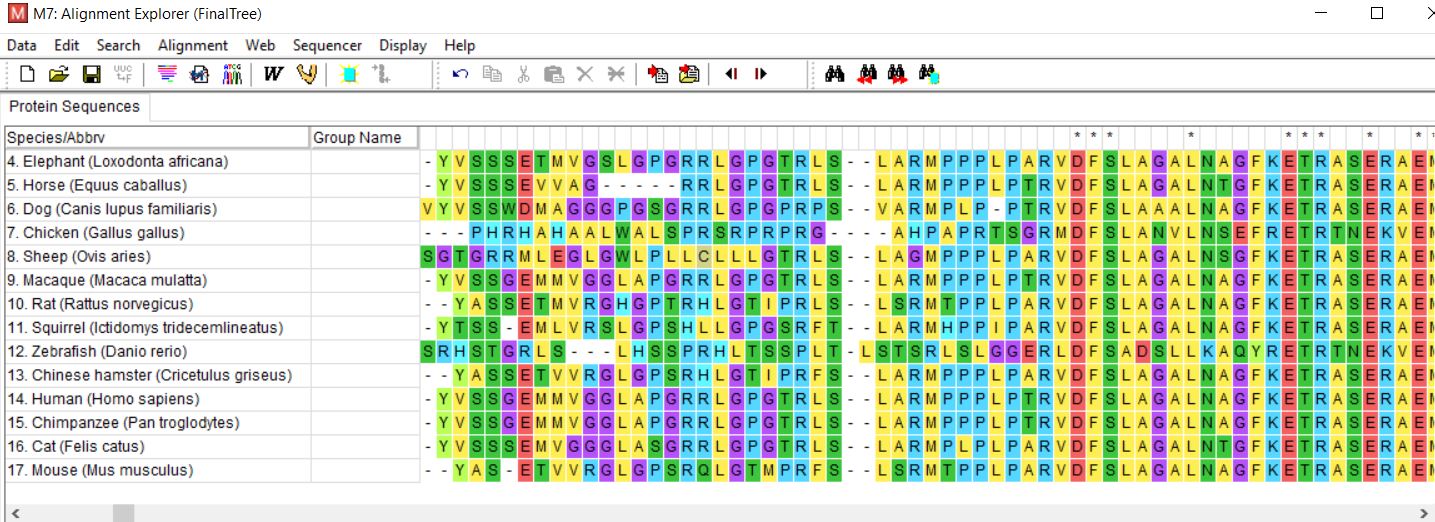
Figure 2: Clustal Omega alignment of GFAP homologs performed in MEGA.
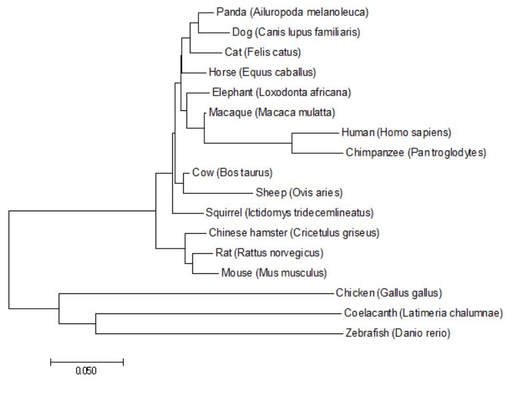
Figure 3: GFAP phylogenetic tree generated using the Neighbor Joining method in MEGA.
What is Maximum Likelihood? The Maximum Likelihood method begins with generating an initial tree based on the Neighbor Joining Method using percent similarity. It then takes this tree and uses a certain statistical model of sequence evolution, such as the Minimum Evolution model. The likelihood models take into account the amount of mutations between nodes, and determine likelihood of these relationships, the more mutations the less likely for the relationship to be true [1,4].
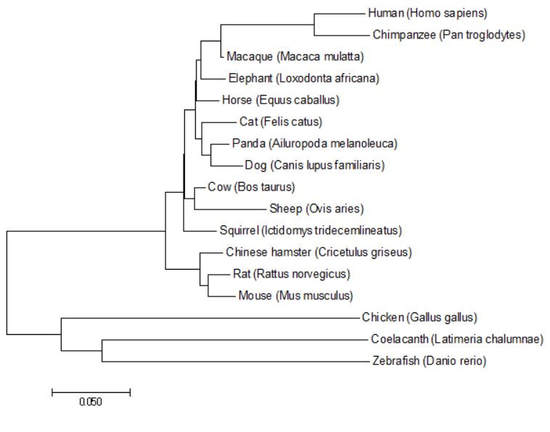
Figure 5: GFAP phylogenetic tree generated using the Minimum Evolution method in MEGA.
DiscussionAll phylogenetic trees that were produced under either the Neighbor Joining, Maximum Likelihood, and/or Average Distance methods showed similar or the same evolutionary structure as one another. They all rationalized that the fish and birds are the most distantly related species, while the monkeys were most closely related to humans for concerning GFAP relatedness. The fact that three of the four trees show the exact same structure gives me confidence in the depicted evolutionary relationships.
|
What is the Neighbor Joining?The Neighbor Joining method is one of the simplest ways to build trees. In the Neighbor Joining method similarity scores are generated by percent identity through protein alignment in Clustal Omega. These similarity scores determine which species are most closely related to one another. Once evolutionary relationships are determined, the Neighbor Joining method then calculates the branch lengths based on how many unique mutations are observed after the species have diverged, with more mutations leading to a longer branch length [2,3].
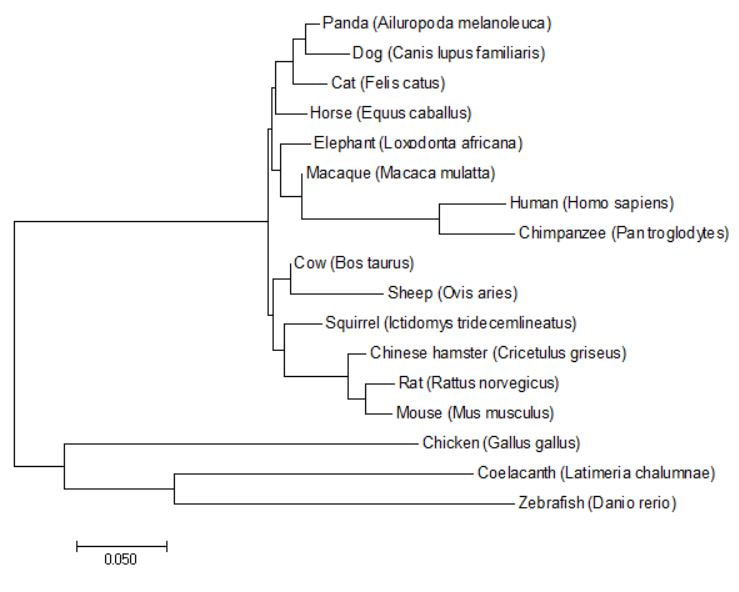
Figure 4: GFAP phylogenetic tree generated using the Maximum Likelihood method in MEGA.
What is Average Distance?The Average Distance method is another simple method for creating phylogenetic trees. This method takes the percent similarity scores to determine which species are most closely related, just like the Neighbor Joining method. The tree is then created with all equal branch lengths, because in the Average Distance method it is assumed that all the species have diverged equally from their most common ancestor [2,4].
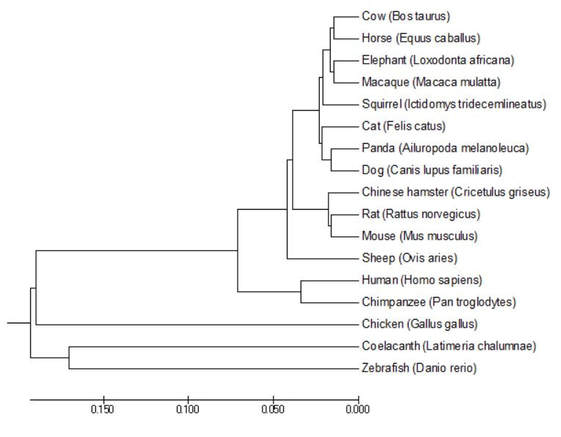
Figure 6: GFAP phylogenetic tree generated using the Unweighted Pair Group Method with Arithmetic Mean (UPGMA) in MEGA.
|
References
[1] Chapter 27: Phylogenetic Reconstruction. (n.d.). Retrieved March 16, 2018, from http://evolution-textbook.org/content/free/contents/ch27.html#ch27-4-2
[2] What is phylogenetics? (2014, December 5). Retrieved March 15, 2018, from https://www.ebi.ac.uk/training/online/course/introduction-phylogenetics/what-phylogenetics
[3] Sievers F, Wilm A, Dineen D, Gibson T, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson J, Higgins D, Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega, MSB 7 (2011) 1-6. PMID: 21988835
[4] Skop, A. (2018). Lab 2: Homology & Phylogeny. Retrieved March 14, 2018, from http://genetics564.weebly.com/homology--phylogeny.html
[2] What is phylogenetics? (2014, December 5). Retrieved March 15, 2018, from https://www.ebi.ac.uk/training/online/course/introduction-phylogenetics/what-phylogenetics
[3] Sievers F, Wilm A, Dineen D, Gibson T, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson J, Higgins D, Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega, MSB 7 (2011) 1-6. PMID: 21988835
[4] Skop, A. (2018). Lab 2: Homology & Phylogeny. Retrieved March 14, 2018, from http://genetics564.weebly.com/homology--phylogeny.html
