This web page was produced as an assignment for Genetics 564, an undergraduate capstone course at UW-Madison.
What is Alexander Disease?
|
Alexander Disease is a rare, fatal, autosomal dominant neurodegenerative disorder that affects the central nervous system [2,7]. Alexander Disease is caused by heterozygous mutations in the gene GFAP, which encodes the glial fibrillary acidic protein, and it predominantly affects infants and children [2,7]. It is part of a larger group of disorders, known as leukodystrophies, which involve the destruction of myelin. Myelin is the fatty coating that insulates our nerve cells and promotes rapid transmission of neuronal signals, which allow our brain to function and communicate. Alexander Disease is also characterized by the presence of “Rosenthal fibers”, cytoplasmic protein inclusions in cells called astrocytes. Astrocytes are the most numerous cell type in the central nervous system, and recent studies on mice have found that the accumulation of Rosenthal fibers in astrocytes correlate with abnormal changes in their phenotype, effecting their normal functions [5].
|

|
Symptoms and Onset of Alexander Disease
Alexander Disease can be categorized into four different types based on age of onset, these types in order are, neonatal, infantile, juvenile, and adult, though previously neonatal was apart of the infantile type [2,6,7]. Variability in phenotype is usually dependent on the age of onset, with the most severe symptoms and faster progression occurring in infants and children, while older juveniles and adults experience a slower progression.
|
Neonatal:
Common onset happens in the first month, and results in severe disability or death within the first two years of life with little variability in symptoms [6]. Symptoms include seizures, hydrocephalus, severe motor disability, severe intellectual disability, and severe white-matter abnormalities in the brain [6]. Juvenile: (22% of affected individuals)
Commonly presents itself between the ages of 4 and 10, with some being diagnosed in their mid-teens. Life expectancy ranges from early teens up to the 20s and 30s due to variability of symptoms as well as slower progression in older juveniles [2,7]. Symptoms can include speech abnormalities, difficulties swallowing, and frequent vomiting, lower limb spasticity, ataxia, slow loss of intellectual function, seizures, megalencephaly, and breathing problems [2,7]. |
Infantile: (42% of affected individuals)
Presents itself within the first two years of life, and affected individuals usually only survive weeks, but some individuals will survive several years, as severity of certain symptoms is variable [2, 7]. Symptoms include progressive psychomotor retardation, frontal bossing and megalencephaly, seizures, hyperreflexia, ataxia, and hydrocephalus [2,7]. Adult: (33% of affected individuals)
Presents itself in adulthood and is highly variable in phenotype. Most cases will see life expectancy to be about 10 to 20 years after presentation, depending on speed of progression and symptom development [3,4,7,8]. Symptoms can include palatal spasms, difficulty swallowing, difficulty speaking, slurred speech, unwanted muscle contraction, hyperreflexia, ataxia, constipation, abnormal sweating, sleep apnea, difficulty walking, and seizures [2,3,4,7,8] |
Diagnosis and Treatment
A diagnosis of Alexander Disease is strongly considered for children who experience one or more of the symptoms listed above, especially megalencephaly, as well as leukodystrophy once other types of leukodystrophy are ruled out [2]. Furthermore, Magnetic Resonance Imaging (MRI) is suggested in order to gain a clear picture of whether or not Alexander Disease is the cause, though genetic testing of a blood sample can give a definitive diagnosis for confirmation of Alexander Disease [2,7].
Currently, there is no a cure for Alexander Disease, but a few supportive treatments are available in order to lessen symptoms, such as certain medicines to help with seizure prevention [7]. Recent research on GFAP in mice has found that mice lacking GFAP might suffer little consequences of having no copies of the gene, indicating a mechanism for turning of GFAP might be able to quell the fatal symptoms in patients in the future [1,2].
Currently, there is no a cure for Alexander Disease, but a few supportive treatments are available in order to lessen symptoms, such as certain medicines to help with seizure prevention [7]. Recent research on GFAP in mice has found that mice lacking GFAP might suffer little consequences of having no copies of the gene, indicating a mechanism for turning of GFAP might be able to quell the fatal symptoms in patients in the future [1,2].
Gene and Protein Function
|
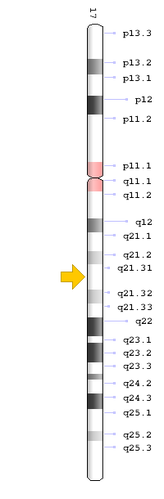
GFAP is an interesting gene that encodes for the glial fibrillary acidic protein, an intermediate filament protein. In humans, the GFAP gene is located on the q arm of chromosome 17 at the position q21.31 [7]. GFAP is a highly specialized gene, because it is primarily expressed in brain cells called astrocytes, unlike other intermediate filament proteins which are expressed in all cells of the body [1]. GFAP has homologs in many species of vertebrates, but is not common among all eukaryotes.
Currently, the exact role of GFAP and its corresponding protein is unknown. Though through recent research and the knowledge that it is an intermediate filament protein, the glial fibrillary acidic protein may play a role in forming the blood-brain barrier, providing mechanical strength, and helping with myelination [1]. Even more interesting, a few studies have found upregulation of GFAP when certain CNS injuries occur, meaning it could play a crucial role in damage control [1]. |
Resources
|
National Organization for Rare Disorders
55 Kenosia Avenue Danbury, CT 06810 Phone: (203) 744-0100 Fax: 203-263-9938 Website: https://rarediseases.org/ |
Waisman Center
1500 Highland Ave Madison, WI 53705 Business Office: (608) 263-1656 Clinics: (608) 263-3301 Director's Office: (608) 263-5940 Website: https://www2.waisman.wisc.edu/scrp-disorders-alex.htm |
United Leukodystrophy Foundation
224 N. 2nd St. Suite 2 DeKalb, IL 60115 Phone: (815) 748-3211 Toll-free: (800) 728-5483 Email: [email protected] Website: http://www.ulf.org/ |
References
[1] Brenner, M. (2014). Role of GFAP in CNS injuries. Neuroscience Letters, 565, 7–13. https://doi.org/10.1016/j.neulet.2014.01.055
[2] Johnson, A. B., & Brenner, M. (2003). Alexander’s Disease: Clinical, Pathologic, and Genetic Features. Journal of Child Neurology, 18(9), 625–632.
[3] Martidis, A., Yee, R. D., Azzarelli, B., & Biller, J. (1999). Neuro-ophthalmic, radiographic, and pathologic manifestations of adult-onset Alexander disease. Archives of Ophthalmology (Chicago, Ill.: 1960), 117(2), 265–267.
[4] Namekawa, M., Takiyama, Y., Aoki, Y., Takayashiki, N., Sakoe, K., Shimazaki, H., … Nakano, I. (2002). Identification of GFAP gene mutation in hereditary adult-onset Alexander’s disease. Annals of Neurology, 52(6), 779–785. https://doi.org/10.1002/ana.10375
[5] Sosunov, A. A., McKhann, G. M., & Goldman, J. E. (2017). The origin of Rosenthal fibers and their contributions to astrocyte pathology in Alexander disease. Acta Neuropathologica Communications, 5, 27. https://doi.org/10.1186/s40478-017-0425-9
[6] Springer, S., Erlewein, R., Naegele, T., Becker, I., Auer, D., Grodd, W., & Krägeloh-Mann, I. (2000). Alexander disease--classification revisited and isolation of a neonatal form. Neuropediatrics, 31(2), 86–92. https://doi.org/10.1055/s-2000-7479
[7] Srivastava, S., & Naidu, S. (1993). Alexander Disease. In M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. Bean, K. Stephens, & A. Amemiya (Eds.), GeneReviews®. Seattle (WA): University of Washington, Seattle. Retrieved from http://www.ncbi.nlm.nih.gov/books/NBK1172/
[8] Stumpf, E., Masson, H., Duquette, A., Berthelet, F., McNabb, J., Lortie, A., … Cossette, P. (2003). Adult Alexander disease with autosomal dominant transmission: a distinct entity caused by mutation in the glial fibrillary acid protein gene. Archives of Neurology, 60(9), 1307–1312. https://doi.org/10.1001/archneur.60.9.1307
Header Image: https://cdna.artstation.com/p/assets/covers/images/001/626/218/large/johan-de-leenheer-neurons-magenta-05-28.jpg?1449707339
[2] Johnson, A. B., & Brenner, M. (2003). Alexander’s Disease: Clinical, Pathologic, and Genetic Features. Journal of Child Neurology, 18(9), 625–632.
[3] Martidis, A., Yee, R. D., Azzarelli, B., & Biller, J. (1999). Neuro-ophthalmic, radiographic, and pathologic manifestations of adult-onset Alexander disease. Archives of Ophthalmology (Chicago, Ill.: 1960), 117(2), 265–267.
[4] Namekawa, M., Takiyama, Y., Aoki, Y., Takayashiki, N., Sakoe, K., Shimazaki, H., … Nakano, I. (2002). Identification of GFAP gene mutation in hereditary adult-onset Alexander’s disease. Annals of Neurology, 52(6), 779–785. https://doi.org/10.1002/ana.10375
[5] Sosunov, A. A., McKhann, G. M., & Goldman, J. E. (2017). The origin of Rosenthal fibers and their contributions to astrocyte pathology in Alexander disease. Acta Neuropathologica Communications, 5, 27. https://doi.org/10.1186/s40478-017-0425-9
[6] Springer, S., Erlewein, R., Naegele, T., Becker, I., Auer, D., Grodd, W., & Krägeloh-Mann, I. (2000). Alexander disease--classification revisited and isolation of a neonatal form. Neuropediatrics, 31(2), 86–92. https://doi.org/10.1055/s-2000-7479
[7] Srivastava, S., & Naidu, S. (1993). Alexander Disease. In M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. Bean, K. Stephens, & A. Amemiya (Eds.), GeneReviews®. Seattle (WA): University of Washington, Seattle. Retrieved from http://www.ncbi.nlm.nih.gov/books/NBK1172/
[8] Stumpf, E., Masson, H., Duquette, A., Berthelet, F., McNabb, J., Lortie, A., … Cossette, P. (2003). Adult Alexander disease with autosomal dominant transmission: a distinct entity caused by mutation in the glial fibrillary acid protein gene. Archives of Neurology, 60(9), 1307–1312. https://doi.org/10.1001/archneur.60.9.1307
Header Image: https://cdna.artstation.com/p/assets/covers/images/001/626/218/large/johan-de-leenheer-neurons-magenta-05-28.jpg?1449707339
Contact Info:
|
|
